Drug metabolism plays a central role in the discovery of new medications. Predicting the metabolic fate of drugs in patients is challenging, yet is essential for minimizing toxicity, maximizing efficacy, and for predicting drug-drug or drug-food interactions. Human cytochrome P450 enzymes (CYPs) are by far the most important players in phase 1 drug metabolism. Together, human CYP3A4 and CYP2D6 contribute to the metabolism of approximately 75% of all drugs in clinical use, and both enzymes are implicated in adverse drug interactions and drug resistance. CYP enzymes catalyze heme-dependent substrate monooxygenation, which typically enhances metabolite lipophilicity and facilitates elimination from the body. The CYP catalytic cycle requires electrons (delivered by a CYP reductase, CPR) in order to activate molecular oxygen for insertion into the substrate.
The catalytic activity of many CYPs is highly sensitive to the identity of the bound substrate ligand, and many CYPs are regulated by allosteric interactions (both with substrate and with other effector molecules). Moreover, the reductase, CPR, is known to undergo large conformational changes as it binds to NADPH (the source of reducing equivalents) and delivers reducing equivalents to the heme center of CYP. Thus, the CYP-CPR system is highly structurally dynamic in ways that are important for CYP function. However, despite the biomedical importance of CYP-CPR, the molecular details of CYP-CPR interactions and their relation to CYP redox catalysis are not well understood.
In collaboration with the Auclair group (McGill Chemistry Department), we have demonstrated that combining HDX-MS with site-specific bioconjugation of allosteric effectors is a powerful means to assess conformational dynamics and allosteric mechanisms in the human CYP3A4. Namely, we have shown that progesterone (a CYP3A4 effector) triggers different allosteric effects (allosteric activation, antagonism, and agonist-like activation) in CYP3A4 depending on the site of bioconjugation. Interestingly, each progesterone conjugate is associated with unique patterns in CYP3A4 structural dynamics as revealed by HDX-MS studies (Figure 4). These studies show that CYP3A4 structure and function are modulated by the identity and location of bound effector ligands and substrates. This conformational plasticity is likely central to the ability of CYP3A4 to metabolize a broad range of substrates. We are currently expanding our efforts to investigate how the CPR enzyme modulates CYP structure and function and to better understand how clinically relevant CPR polymorphisms affect drug metabolism by CYP.
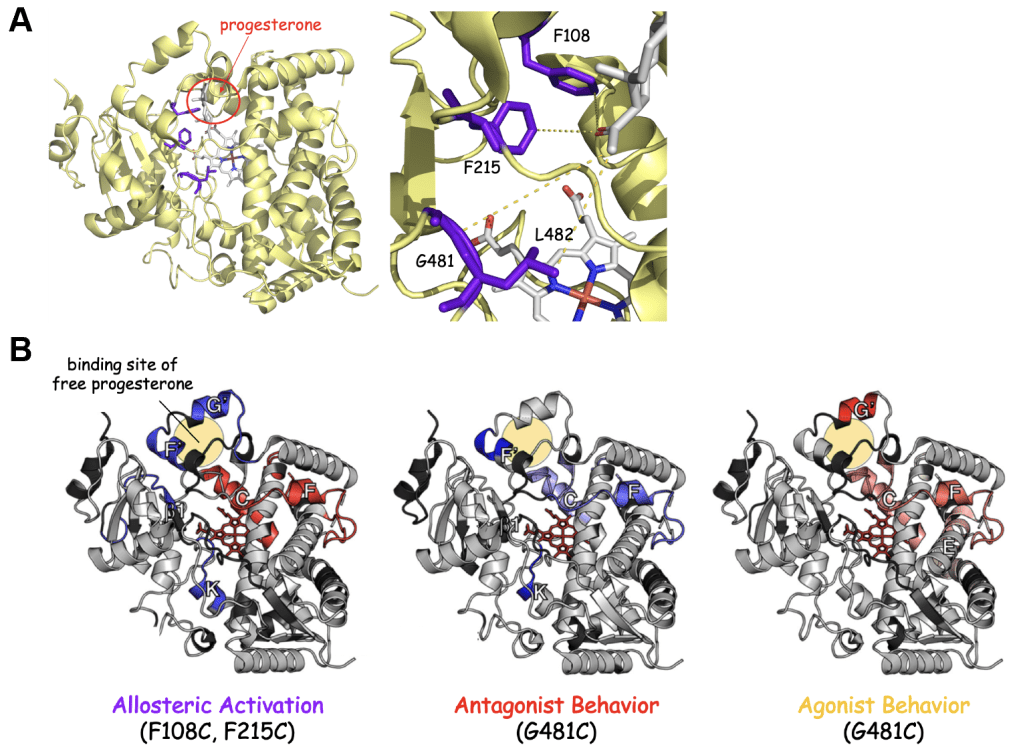
Figure 4. Site-specific conjugation of progesterone leads to distinct perturbation of CYP3A4 dynamics. A) Structure of CYP3A4 showing the locations where the progesterone allosteric effector was conjugated. B) HDX dynamics of CYP3A4 differ depending on the site of progesterone conjugation. The F108C, F215C and G481C conjugates undergo organization in the putative allosteric site (beige circle), consistent with binding of the conjugated progesterone. The F108C and F215C enzymes are allosterically activated by progesterone conjugation, while the G481C conjugate is inhibited. These differential effects on CYP3A4 catalysis could be related to the differential effects of progesterone conjugation on the dynamics of the F and C helices, which harbor catalytic residues and interact with the P450 reductase, respectively. In support of this, the G481C conjugate, which is also activated by progesterone conjugation also undergoes a relaxation the F and C helices – mimicking the F108C and F215C conjugates. The allosteric site of the G481C conjugate (helices F’ and G’) remains dynamic, however, suggesting that the conjugated progesterone moiety is binding to an alternative (agonist) site.
